Compsci 100, Spring 2009, DNA Splicing
You should snarf assignment dna from
http://www.cs.duke.edu/courses/spring09/cps100/snarf or browse the
code directory for code files provided. See the dna howto file for help and more instructions.
Genesis of Assignments Linked to DNA/Genomics
Rich Pattis created an
assignment based on the whole genome shotgun reassembly algorithm as
part of the Nifty Assignments
Archive, but there's nothing in the archive about the assignment and
he hasn't used it in a while. We used
that assignment at Duke in Compsci 4G for several
years.
This current assignment was developed in 2002, used for two years, then
resurrected in the Spring of 2008 and used since then. It's turned into
a very different assignment over that time. These differences leverage
discussion of tradeoffs more than the original assignment, and were
motivated in part by opportunities (and differences) provided by Java
compared to C++. The assignment is meant to illustrate pragmatically the
benefits of using a linked list.
Background on DNA, Restriction Enzymes, and PCR
This background is interesting, but not really needed to do the
assignment. There are some good stories here, but if you want to
get to the assignment, you can skip this stuff.
Kary Mullis, the inventor of PCR, is an interesting character. To see
more
about him see this archived copy of a 1992
interview in Omni Magazine,
this 1994
interview as parf of virus myth,
his personal website which includes
information about his autobiography Dancing Naked in the Mind Field, though you can read this free
Nobel autobiography as well.
What You do for This Assignment
In this assignment you're given a class that represents a strand of DNA
and code to repeatedly cleave/cut and join new DNA into the existing
strand --- this process is explained conceptually
below -- your code models the process. You'll experiment with the
existing strand class and then create a new strand class that's much
more efficient in simulating cleaving/cutting/joining DNA. You'll run
experiments to understand the tradeoffs in the implementation of these
classes.
In particular you'll need to do three things.
- Benchmark the code in
SimpleStrand.cutAndSplice that uses
String.split to find all occurrences of an enzyme and
replaces the enzyme with another strand of DNA. The benchmarking is
done by the class DNABenchMark and is explained in more detail
below. You'll run virtual experiments in code to show two things:
-
First, show that the algorithm/code
in
SimpleStrand.cutAndSplice
is O(N)
where N is the size of the recombined strand
returned. In your README describe the results of the process and
reasoning you use to demonstrate the O(N). You
should have empirical results and timings that demonstrate the O(N) behavior. Graphing the data can be useful in
showing behavior.
-
Second, determine on your machine the largest
splicee string (the string spliced into the DNA strand)
that works without generating a Java Out Of
Memory error when run using the -Xmx 512M heap-size
specification (see the howto). In determining
this largest string the code you use, given in
DNABenchMark
should only use strings whose lengths are a power of
two. Report
this result in your README by providing the power-of-two string
you can use without running out of memory with the input file
ecoli.dat which has 646 cut points in an original strand of
4,639,221 base-pairs with the restriction enzyme EcoRI: "gaattc".
Describe how you determined this result and how
long your program takes on your machine to create the recombinant strand
is constructed using a splicee string
whose length is the greatest (power of 2) that can be used to create
the recombinant cut-and-spliced strand before
memory is an issue. If you double the size of the heap available to the
Java runtime (-Xmx1024M) does your machine support the next
power-of-two strand (i.e., the one that couldn't run with -Xmx1024m)?
If so, how long does it take with ecoli.dat. You should also
run with -Xmx2048M if your machine has enough memory to support
this.
Be sure to report on running with consecutive powers of two for both
-Xmx arguments and size-of-string. If your machine doesn't support
-Xmx512M start with 256M instead.
- Here's the output of running
DNABenchMark on the data
file ecoli.dat
dna length = 4639221
SimpleStrand: splicee 256 time 0.314 before 4,639,221 after 4,639,221 recomb 4,800,471
SimpleStrand: splicee 512 time 0.253 before 4,639,221 after 4,639,221 recomb 4,965,591
SimpleStrand: splicee 1,024 time 0.235 before 4,639,221 after 4,639,221 recomb 5,295,831
SimpleStrand: splicee 2,048 time 0.253 before 4,639,221 after 4,639,221 recomb 5,956,311
SimpleStrand: splicee 4,096 time 0.254 before 4,639,221 after 4,639,221 recomb 7,277,271
SimpleStrand: splicee 8,192 time 0.287 before 4,639,221 after 4,639,221 recomb 9,919,191
SimpleStrand: splicee 16,384 time 0.375 before 4,639,221 after 4,639,221 recomb 15,203,031
SimpleStrand: splicee 32,768 time 0.498 before 4,639,221 after 4,639,221 recomb 25,770,711
SimpleStrand: splicee 65,536 time 0.784 before 4,639,221 after 4,639,221 recomb 46,906,071
Exception in thread "main" java.lang.OutOfMemoryError: Java heap space
at java.util.Arrays.copyOf(Arrays.java:2882)
at java.lang.AbstractStringBuilder.expandCapacity(AbstractStringBuilder.java:100)
at java.lang.AbstractStringBuilder.append(AbstractStringBuilder.java:390)
at java.lang.StringBuilder.append(StringBuilder.java:119)
at SimpleStrand.cutAndSplice(SimpleStrand.java:58)
at DNABenchMark.strandSpliceBenchmark(DNABenchMark.java:77)
at DNABenchMark.main(DNABenchMark.java:109)
-
You must design, code, and test
LinkStrand that implements
the IDnaStrand interface so that your implementation
passes the JUnit tests in TestStrand. See the DNA howto for help with JUnit. Passing
these tests doesn't guarantee full credit since the tests are
about correctness, not about efficiency. Your code should avoid
recalculating values that can be determined by other means. In
particular the length of a LinkStrand strand should
be calculated in O(1) time once the
strand is created. Implementing LinkStrand is the
bulk of the coding work for this assignment. You'll need to
implement every method and use the JUnit tests to help determine
if your methods are correctly implemented.
- You must run virtual experiments to show that your linked-list
implementation is O(B) for a strand with B breaks as described below. To use
LinkStrand in the benchmarking code, change the
string
"SimpleStrand" to "LinkStrand" in the
main
method of the class
DNABenchMark class.
For example, note that
there
are 646 breaks for ecoli.dat when
using EcoRI "gaattc" as the restriction enzyme. You'll need to make many
runs to show this, not just one.
Describe in your README how you show this
and make sure your submitted code supports your experiments and
conclusion.
Restriction Enzyme Cleaving Explained
You'll need to understand the basic idea behind restriction enzymes to
understand
the simulation you'll be writing, modifying, and running.
Restriction enzymes cut a strand of DNA at a specific location, the
binding site, typically separating the DNA strand into two
pieces. In the real chemical process a strand can be split into several
pieces at multiple binding sites, we'll simulate this by repeatedly
dividing a strand.
Given a strand of DNA "aatccgaattcgtatc" and a restriction
enzyme like EcoRI "gaattc", the restriction enzyme locates each
occurrence of its
pattern in the DNA strand and divides the strand into two pieces
at that point, leaving either blunt or sticky ends as described
below. In the simulation there's no difference between a blunt and
sticky end, and we'll use a single strand of DNA in the simulation
rather than the double-helix/double-strand that's found in the
physical/real process.
Restriction enzymes have two properties or features: the pattern of DNA
that marks a site at which separation occurs and a number/index
that indicates how many characters/nucleotides of the pattern
attach to the left-part of the split strand.
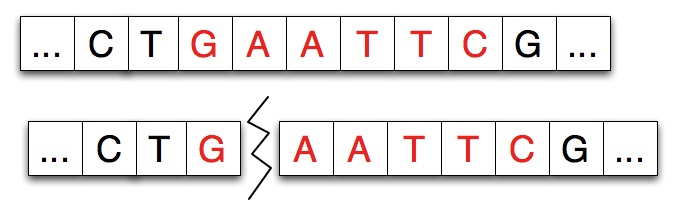
For example, the diagram below shows a strand split by EcoRI.
The pattern for EcoRI is "gaattc" and the index of the split is
one indicating that the first nucleotide/character of the restriction
enzyme adheres to the left part of the split.
In some experiments, and in the simulation you'll run, another strand
of DNA will be spliced into the separated strand. The strand spliced in
matches the separated strand at each end as shown in the diagram below
where the spliced-in strand matches with G on the left and
AATTC on the right as you view the strands.
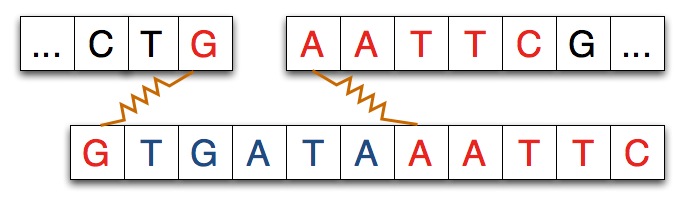
When the spliced-in strand joins the split strand we see a new,
recombinant strand of DNA as shown below. The shaded areas indicate
where the original strand was cleaved/cut by the restriction enzyme.

Your code will be a software simulation of this recombinant process: the
restriction enzyme will cut a strand of DNA and new DNA will be
spliced-in to to create a recombinant strand of DNA. In the
simulation the code simply replaces every occurrence of the restriction
enzyme with new genetic material/DNA --- your code models the process
with what is essentially string replacement.
Simulation/Alternate Implementations
The code below finds all occurrences of a restriction enzyme
like "gaattc" and splices in a new strand of DNA, represented
by parameter splicee to create a recombinant strand. The
strand spliced-in replaces the enzyme. In the simulation
the enzyme is removed each time it
occurs/finds a binding site. The characters representing
the enzyme are replaced by splicee. (This simulates the
process of splitting the original DNA by the restriction enzyme,
leaving sticky/blunt ends, and binding the new DNA to the split
site. However, in the simulation the restriction enzyme is removed.)
This code is from the class SimpleStrand you're given. The
String representing DNA, which is myInfO, is split at every
occurrence of the string parameter enzyme. The spaces are
added before the split as shown below to ensure that characters
representing the enzyme that are found at the beginning or ending
of the DNA are found as part of calling
.split(enzyme).
public IDnaStrand cutAndSplice(String enzyme, String splicee){
String toSplit = " "+myInfo+" ";
String[] all = toSplit.split(enzyme);
if (all[0].trim().equals(myInfo)){
return EMPTYSTRAND;
}
StringBuilder recombined = new StringBuilder(all[0]);
for (int k = 1; k < all.length; k++) {
recombined.append(splicee);
recombined.append(all[k]);
}
return new SimpleStrand(recombined.toString().trim());
}
As the spliced-in strand splicee grows in size the code
above will take longer to execute even with the same original strand of
DNA and the same restriction enzyme. Creating the recombinant strand
using the code above is an O(N) operation
where N is the size of the resulting,
recombinant strand (you have to justify this in your README).
As part of this assignment you must develop an alternate implementation
of DNA, rather than a simple String, that makes the complexity of the
splicing independent of the size of the spliced-in strand. Each splice
operation, simulated by the call to append above, should be
O(1) rather than O(S) for a splicee strand with S
characters/base-pairs. In your new implementation, the complexity of
creating the recombinant strand will be O(B) where B is the number
of breaks/splits created by the restriction enzyme. For a recombinant
strand of size N where N
>> B (>> means much bigger than) this is
significantly more efficient both in time and (especially) memory.
Implementation Specifics
You'll be developing/coding a class LinkStrand that implements
a Java interface IDnaStrand. The class
simulates cutting a strand of DNA
by a restriction enzyme and appending/splicing-in a new strand. The code
supplied with this project gives specifics as to the interfaces and the
howto for this assignment also supplies more
details.
You must use a linked-list to support the operations -- specifically the
class LinkStrand should maintain pointers to a linked list
used to represent a strand. You should keep and maintain a pointer to
the first Node of the linked list and to the last node of
the linked list. These pointers are maintained as class
invariants -- they are true after any method in the class executes
(and thus before any method in the class executes).
A Strand of DNA is initially representing by a linked list with one
Node, the Node stores one string representing the entire strand of
DNA.
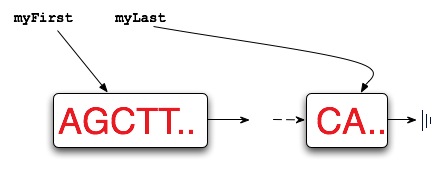
Every linked list representing DNA maintains pointers to the first and
last nodes of the linked list. Initially, before any cuts/splices
have been made,
both myFirst
and myLast point to the same node since there is only
one node even if it contains thousands of characters
representing DNA. The diagram below shows a list with at
least two nodes in it. If any nodes are appended, the value of
myLast must be updated to ensure that it correctly points
to the last node of the new list.
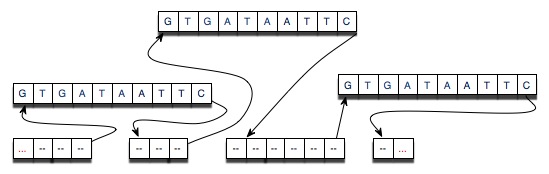
The diagram below shows the results of cutting an original strand of DNA
at three points and then splicing-in the strand "GTGATAATTC" at
each of the locations at which the original strand was cut.
Linked List Details
Grading
Think of this assignment as being worth 40 points.
DNA Simulation Grading Standards
| description
| points
|
| Benchmark code and explanation for O(N)
| 10 points
|
LinkStrand that works as described: both correctly and
efficiently.
| 20 points
|
| Benchmark code and explanation for O(B)
in creating recombinant strands.
| 10 points
|
Submit your project using the assignment name linked-dna.
Extra info about this assignment:
{kind=link}